FAFDrugs4

Partial declines in the productivity of pharmaceutical R&D departments threaten the sustainability of the current business model [6-7]. During many years, the drug discovery process involved chemical synthesis and in vivo testing with optimization of the compounds pharmacokinetic, metabolic and toxic properties postponed to later stages.
In recent years there has been increasing awareness about the importance of predicting and optimizing the Absorption, Distribution, Metabolism, Excretion and Toxicity (ADME-Tox) properties of chemical compounds along the discovery process rather than at the final stages. Indeed, a study in the 1990s showed that several reasons could explain why drugs were failing in development. At that time, new chemical entities were essentially dropped because of poor pharmacokinetic properties, lack of efficacy and toxicity [8-9].
But, the reasons for failures are still manifold: wrong target, poor pharmacokinetics, animal toxicity, lack of clinical efficacy, drug-drug interaction with other drugs, commercial reasons, formulation issues and adverse reactions in humans [10]. Recent analysis suggests that over 90% of failures are now due to toxicity, with hepatotoxicity and cardiovascular implications alone causing two out of three market withdrawals [11].
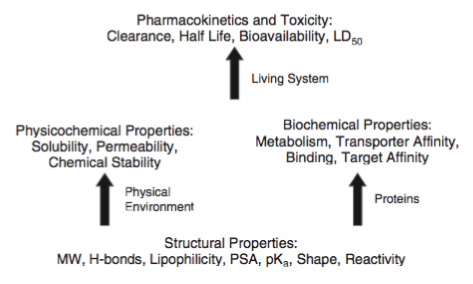
To assist the process, several in vitro and in silico approaches have been devised to for instance predict some key ADME-Tox properties [12], and, in this context and with regard to computer prediction methods, the well-known “rule-of-five” can be seen as a milestone of in silico models, it comprises a set of rules that attempts to predict if a molecule could be administered orally [13]. A clear observation at this time was molecules with an increased molecular weight and lipophilicity were added to the HTS screening collections. The approach has been re-visited numerous times and the overall output of these new investigations supports Lipinski findings, although anti-infectious drugs, some anti-cancer drugs and natural compounds tend to escape these rules [14]. Although this kind of analysis and concepts about compound library profiling are still under debates, several studies suggest that a paradigm shift in drug discovery has occurred. One key challenge is to include in drug discovery campaigns (high-throughput and in silico screening) and whenever appropriate (depending on the target/disease types, potency, selectivity, stage of the project ...) the right ADME/Tox property filtering. In addition, it is critical to simultaneously optimize binding affinity/selectivity, pharmacokinetic properties while avoiding toxicity. Prediction tools can thus assist both. It is of course important to find a balance between removing basically all molecules and retaining all. This is one of the reasons why we have an intermediate list where molecules are only flagged and not added to the rejected list.
Here, we offer an online service called FAF-Drugs4, with optimized capabilities as compared to the earlier FAFDrugs3 version of the package [1], hosted on the RPBS platform and managed by the Mobyle Portal [16]. The aim is to help drug designers and medicinal chemists to prepare compound libraries prior to virtual/experimental screening and/or to assist decision-making prior to synthesis or ordering compounds...