How to use the FAF-Drugs4 web-server.
This page describes how to use and run FAF-Drugs4 hosted on the Mobyle Portal.
» Accessing to the service.
- Go to Mobyle Portal.
- In the left panel, select in the Programs Section » Drugs » FAF-Drugs4 » FAF-Drugs4
- Optionally, at the top right, sign in your email to be notified when your job is launched and finished.
Additionnaly, you can register for an account, which enables you to store your bookmarked data and results on the server, without any time limit.
You can also connect using the same e-mail and password from any place.
- » Optional Services :
- In the » FAF-Drugs4 Section, you can see three additional services, namely Bank-Cleaner, Bank-Formatter and Filter-Editor.
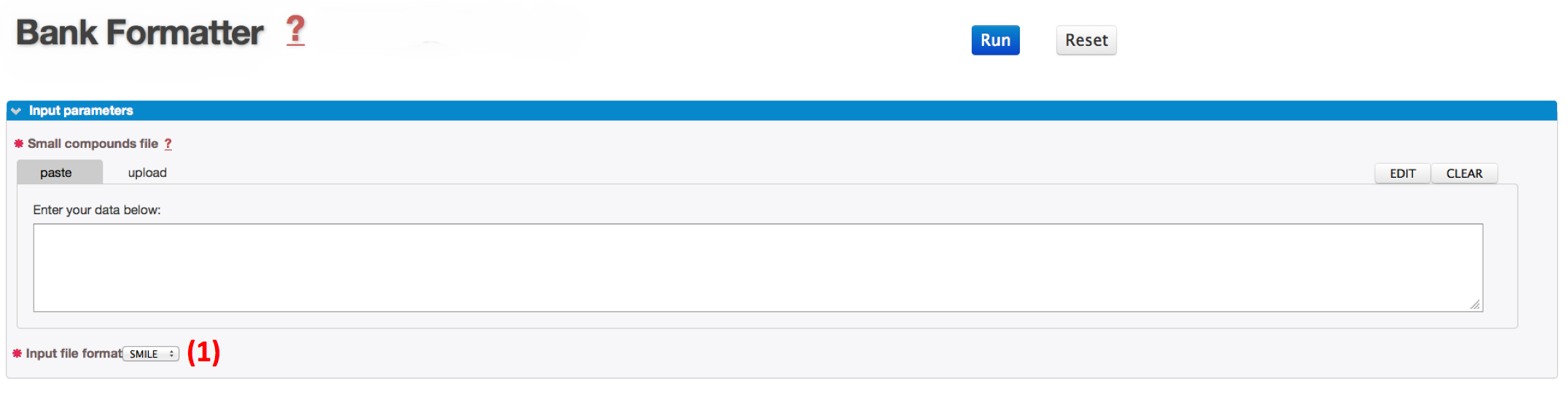
• Bank-Formater is a tool for preparing your compound library in order to properly process the job, especially regarding the Symyx SDF file format recommandations.
It takes SMILES and SDF file formats as input, and generate exclusively an acceptable SDF file.
In order to launch the process, you need to paste or upload your own file.
Before running the program by clicking on the RUN button, be sure that you had selected the file format of the input file (1).
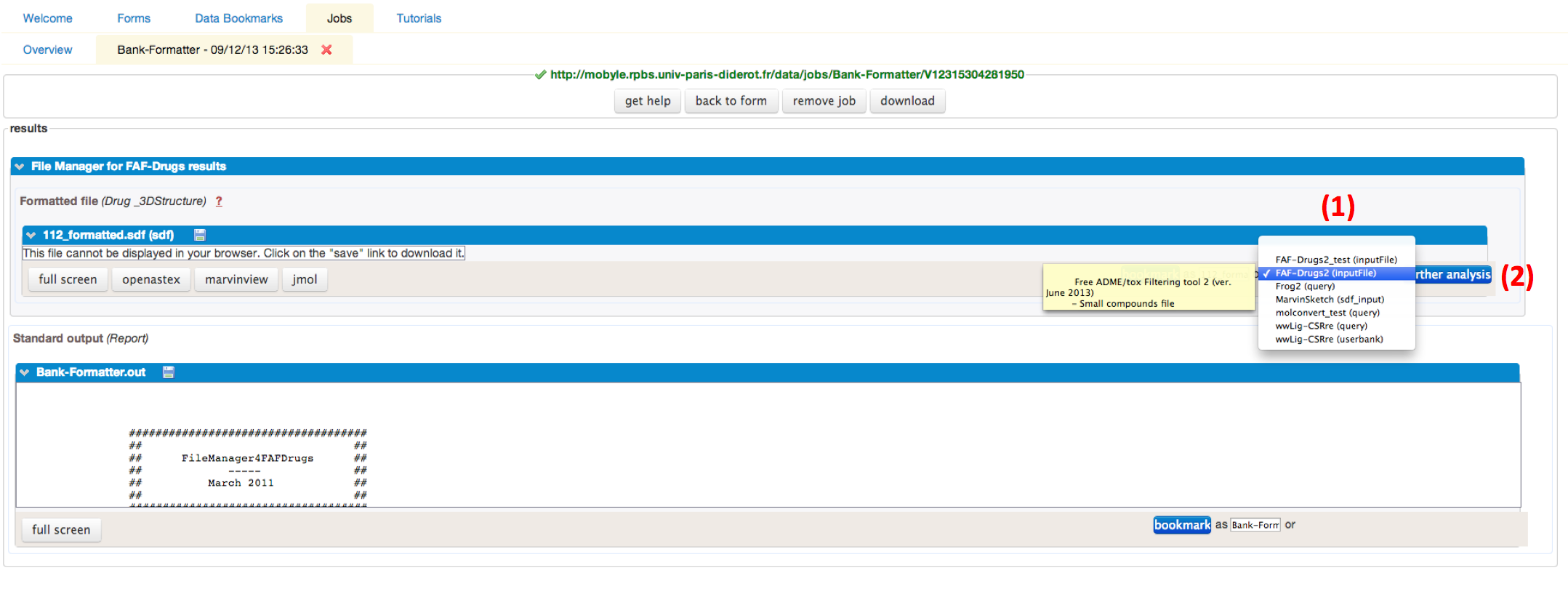
When the process is finished, the SDF file generated can be automatically redirect, as input file, to the FAF-Drugs4 service by using the drop-down menu (1), select FAF-Drugs4 (input file), and click on the further analysis (2) button
• Bank-Cleaner is a tool derived from FAF-Drugs4 only dedicated for the data curation. It uses the FAF-Drugs4 procedure without filtering. To understand how it works, please go to see the FAF-Drusg4 flow chart section.
In order to launch the process, you need to paste or upload your own file. If your file is a SMILES input, you can use the Bank-Formatter tool.
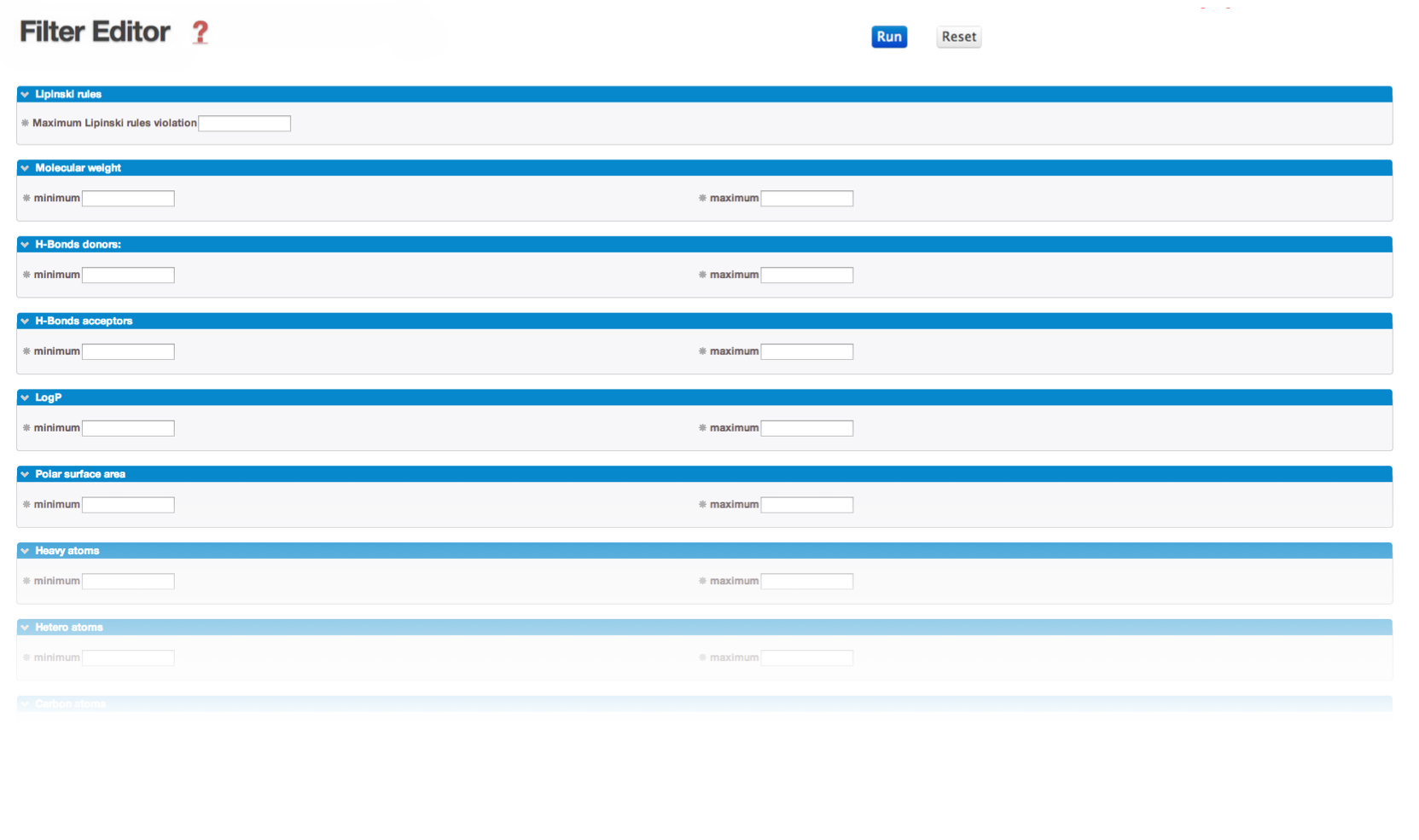
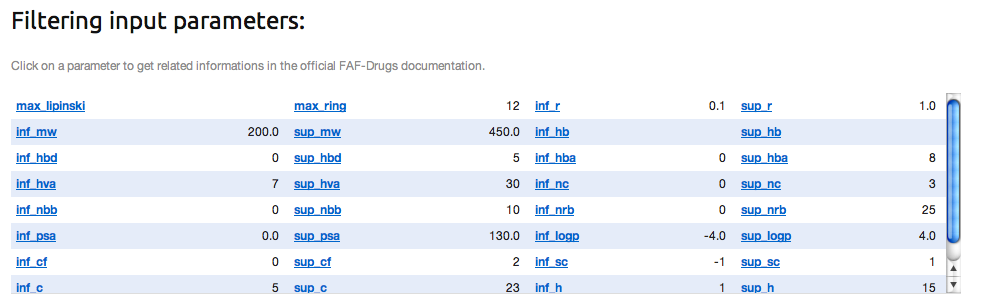
» Filter-Editor is a tool to design your own filtering parameter file. To this end, you can enter your parameters in the dedicated several areas, and click on the RUN button:
It will automatically update the internal standard parameter file with your own filtering thresholds.
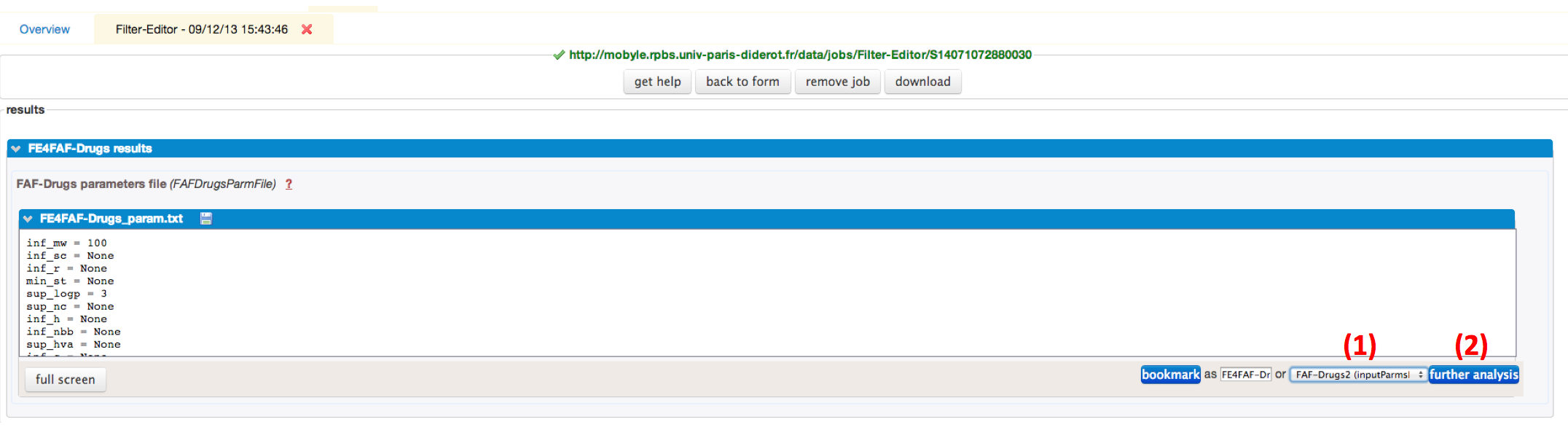
To continue the FAFDrugs4 process with customized parameters, you need to use the drop-down menu (1), select FAF-Drugs4 (inputParmsFile), and click on the further analysis (2) button.
As you can see, your values that will customizing the filtering ranges are displayed.
» Testing the service.
- You can test FAFDrugs4 by using an embedded demonstration mode.
- If you click on the corresponding radio button (1), and running the program (2), a small internal SDF file containing 9 compounds will be processed.
r
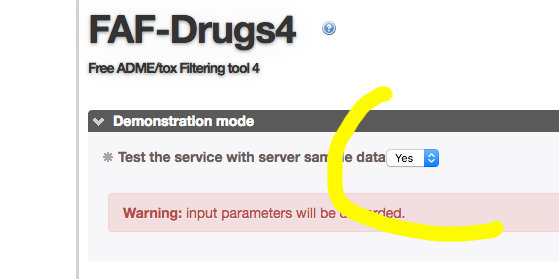
- WARNING !! : If you have tested FAF-Drugs4 before running your own compounds, be sure you have deselected the testing (1) radio button !!
» Input Files, Options and Parameters.
Note: According to MDL Information Systems - Symyx Technologies Structure Data Files guidelines, compounds collection must be in a valid SDF file [17].
It means that each header block of molecules must be composed by 3 lines, the first one must contain the molecule name or the ID tag in a one continuous pattern.
OpenBabel works upon that guidelines, so if no name/ID is specified, you must perform this adjustment.
To this end, you must firstly check if your input file is in an
acceptable format, and if needed, you can use the Bank-Formater tool previously described.
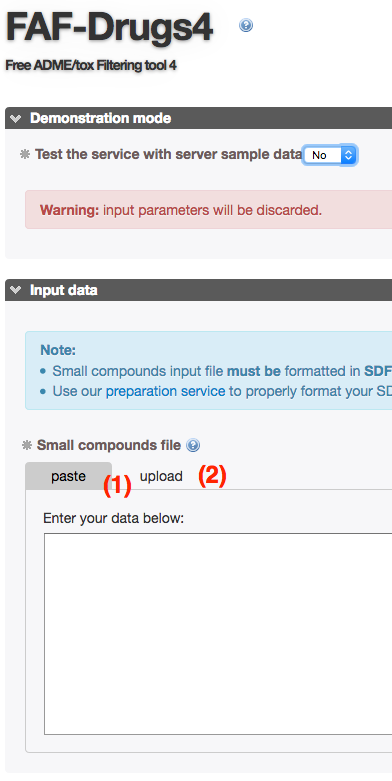
- You have 2 possibilities to enter your data, both of these being selected by clicking on the corresponding radio button:
- (1). Copy/Paste: Simply copy and paste your SDF file in the text area.
- (2). Local file: Select a local file stored on your computer and upload it.
- Several filtering options are available:
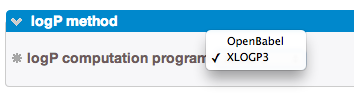
- 1. You can choose which method will compute the logP values between the embedded logP method of OpenBabel [3] or the XLOGP3 program under license agreement established between our lab and Pr R. Wang [5].
- 2. In-House or published Physico-Chemistry filters:
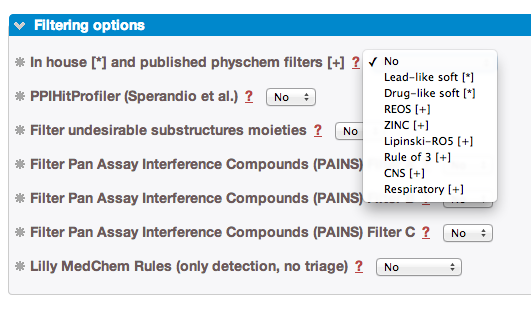
- You can access it by using the drop-down menu to differents filters, ranges are detailed on the filter dedicated page:
» REOS filter, according to Walters et al. [33].
» ZINC filter, according to Irwin et al. [24].
» Lipinski RO5 filter, fitting the Lipinski's of thumb.
According to Lipinski et al. [13], that filter is only active on 4 descriptors: logP, Molecular Weight, H-Bond Donors and Acceptors.
» Rule of 3 filter, according to Congreve et al. [35]. That filter could be useful when constructing fragment libraries for efficient lead discovery. It involves logP, Molecular Weight, H-Bond Donors and Acceptors, number or rotatable bonds and tPSA.
» CNS filter, according to Jeffrey et al. [36], which give some filtering guidelines in order to selecting only the compounds that could enter the central nervous system.
» Inhaled - Respiratory filter, according to Ritchie et al. [25], which give some filtering guidelines in order to selecting only the compounds that could be nasal/inhalatory bioavailable.
- 3. Then, you can apply, or not specifics options:
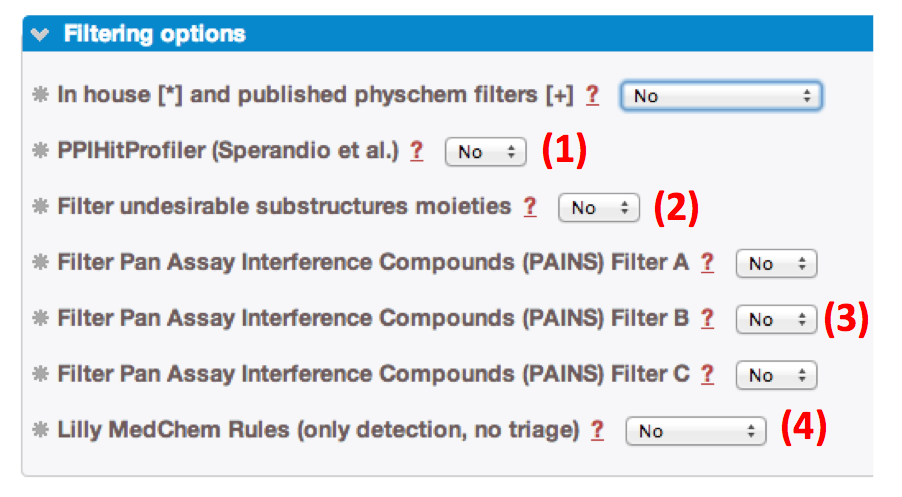
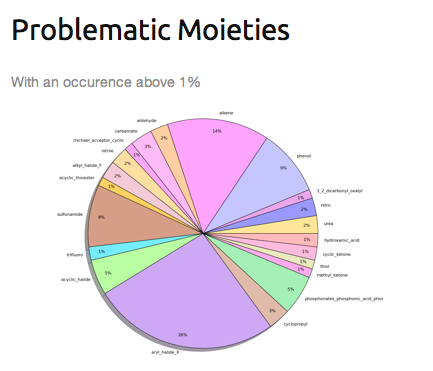
» Filtering compounds with undesirable moieties (2). Functionnal groups detected are detailled in the Toxicophores section.
» Detecting and filtering PAINS (3). The detection of the 480 Pan Assay INterference compoundS (PAINS) according to Baell et al [23], is documented in the PAINS section.
» Applying Lilly MedChem Rules (4). Warning, in term of compound profiling, only detection is computed according to Bruns et al [53]. Please, note that no compounds are filtered upon these rules.
» Running the service.
- You can now run FAFDrugs4 by clicking on the Run button (1).
- Clicking on the Reset (2) button will clear all input data you entered, and stop the process.
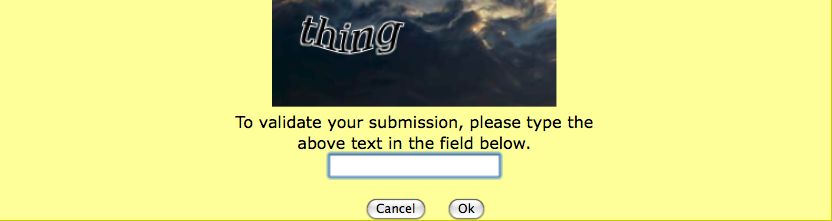
The first time you will run FAF-
Drugs4, you will have to enter a security string. Don't worry, you only have to do this the first time you run FAF-
Drugs4.
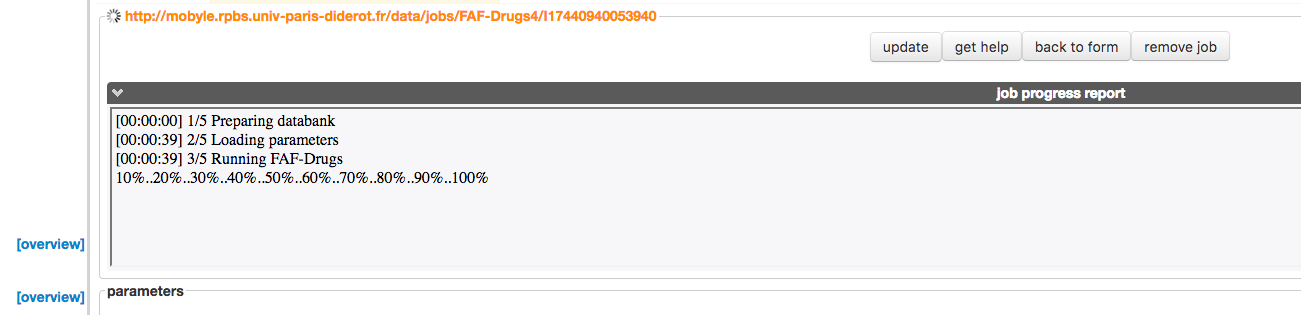
- Once you have submitted your data, a new job will be created, and you will be redirected to this new job tab page as shown bellow.
Two possibilities are there:
» The job is pending and you have to wait until it will be launched.
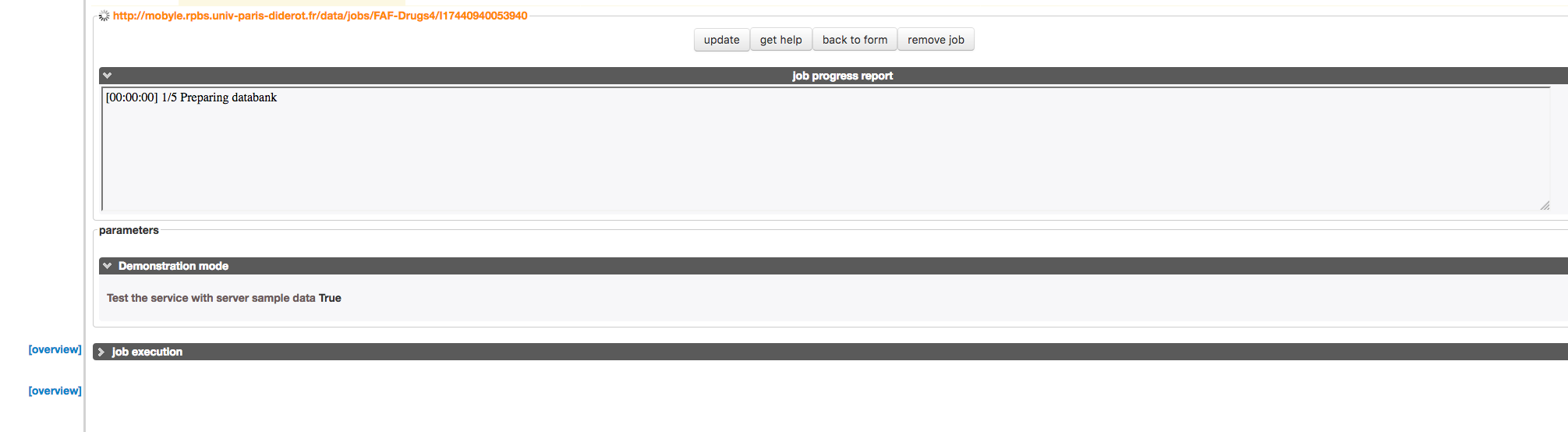
» The job is running and you just have to wait for results to appear.
At that stage, if you have previously sign in your email, you may be notified by an email message that your job was launched and is now running.
You will receive another email message when the computations are finished.
- You can already see the process status in the dedicated progressing window (1).
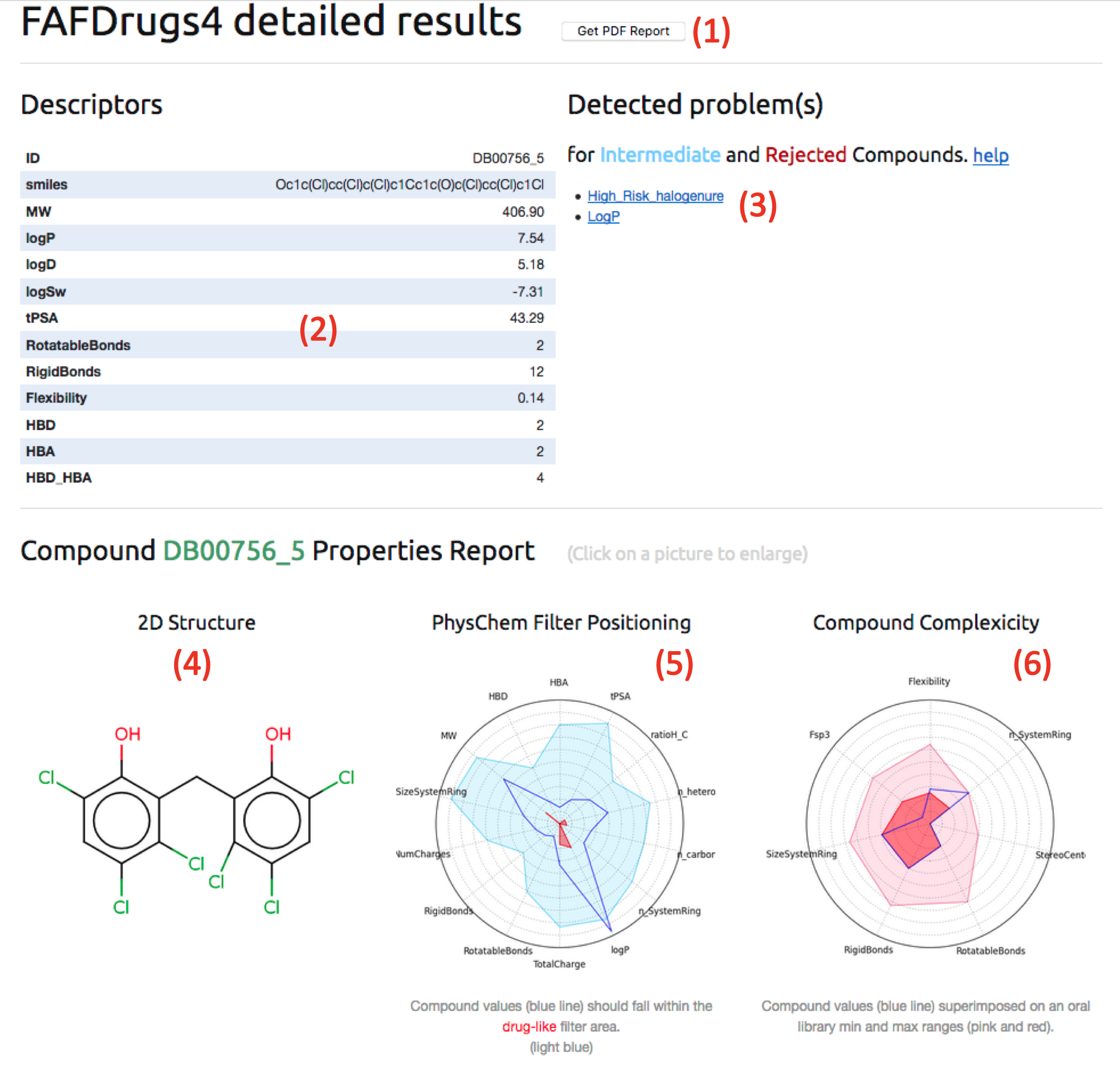
» Output Description.
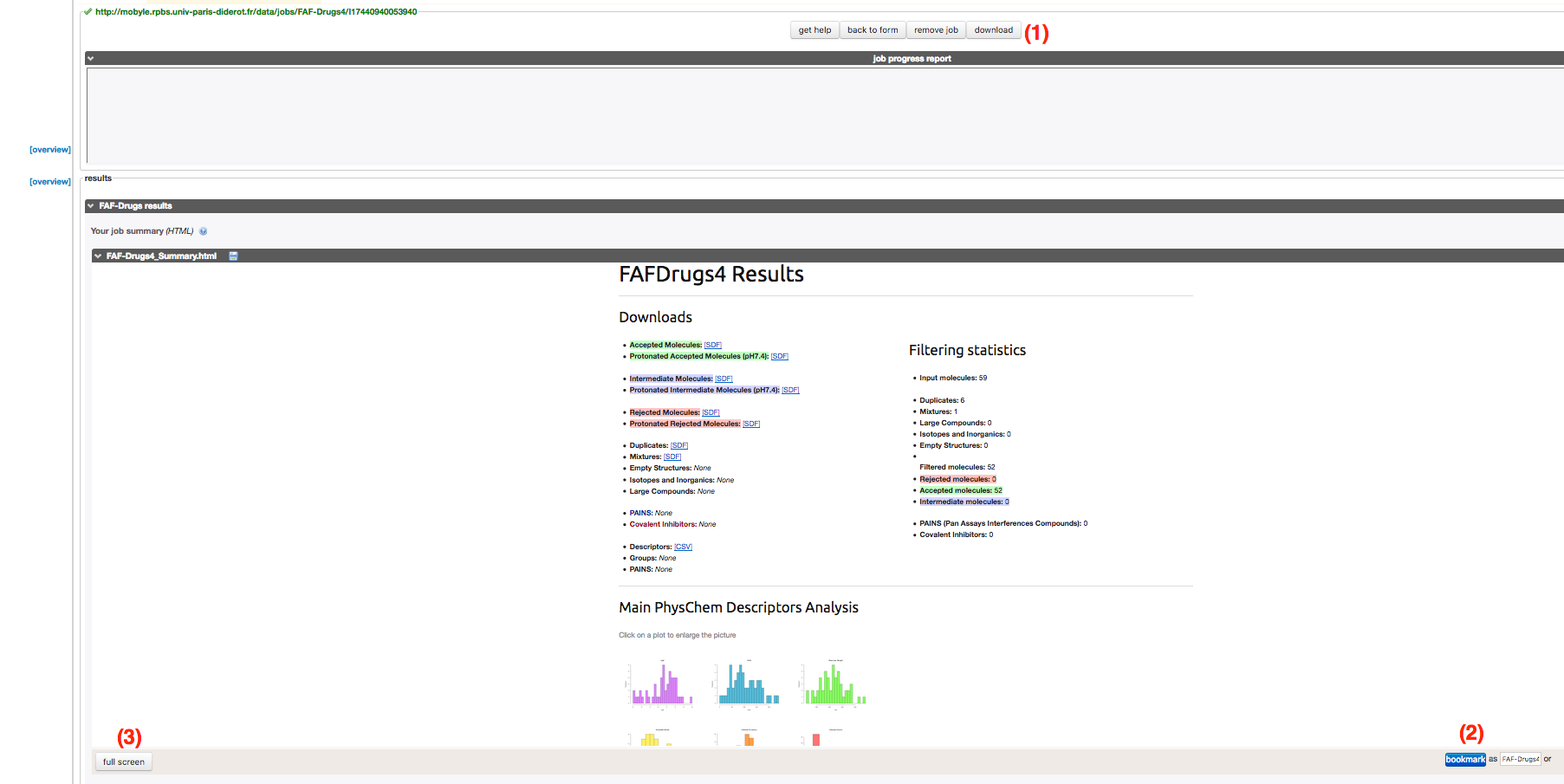
- In Mobyle, once your job is finished, you will be redirected to the mobyle results page. This results page is shown on the picture below.
Differents results pages are actually contained in several files and all can be downloadable on your own computer using the Download button (1).
You also have the possibility to bookmark the result page using the Bookmark button (2) (enter the bookmark name in the field on left), in order to access to the results at some other time.
The web-based can be available by clicking on the full screen view button (3) to reach the following free-handly graphical web-based interface.
- Before developping the “web-based“ way, as explain before on the Mobyle results page you will find several windows (each time associated with a file that can be saved on your computer):
- 1. The links for downloading the ending filtering files (Accepted.sdf, Intermediated.sdf and Rejected.sdf).
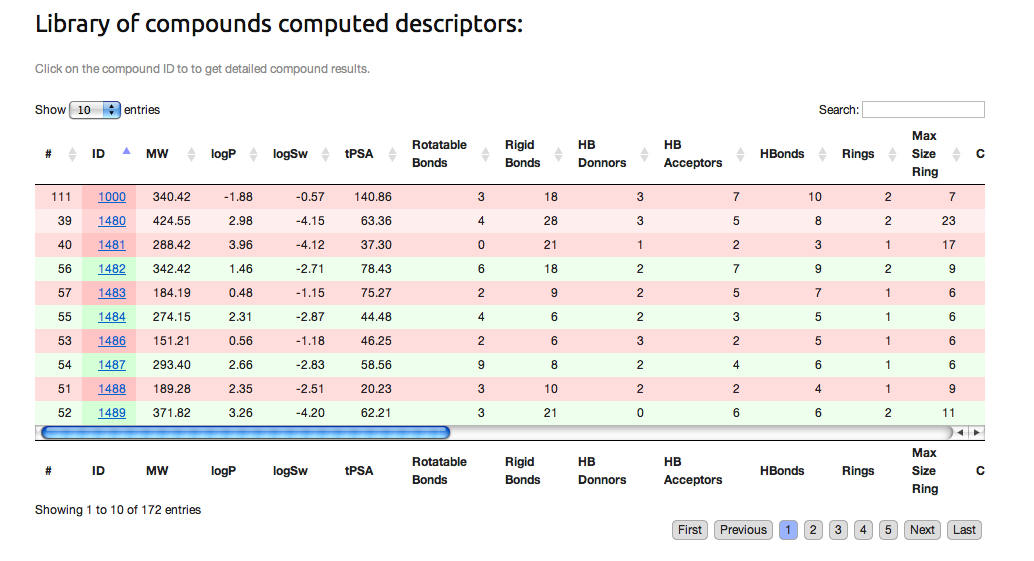
- 2. The "results.csv" file which gather all physico-chemistry results of the filtering process.
- 3. The "groups.csv" file which gather all the results of the substructures/undesirables moieties searching process ( if the dedicated option was activated)
- 3. The "pains.csv" file which gather all the results of the PAINS searching process ( if the dedicated option was activated)
- 4. A simple window summarized the filtering process.
- 5. The standard output of FAFDrugs4 (i.e.terminal display)
- 6. Each separate pictures wich are displayed in the “web-based“ version.
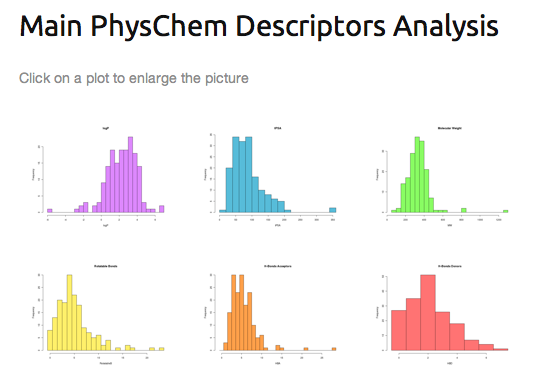
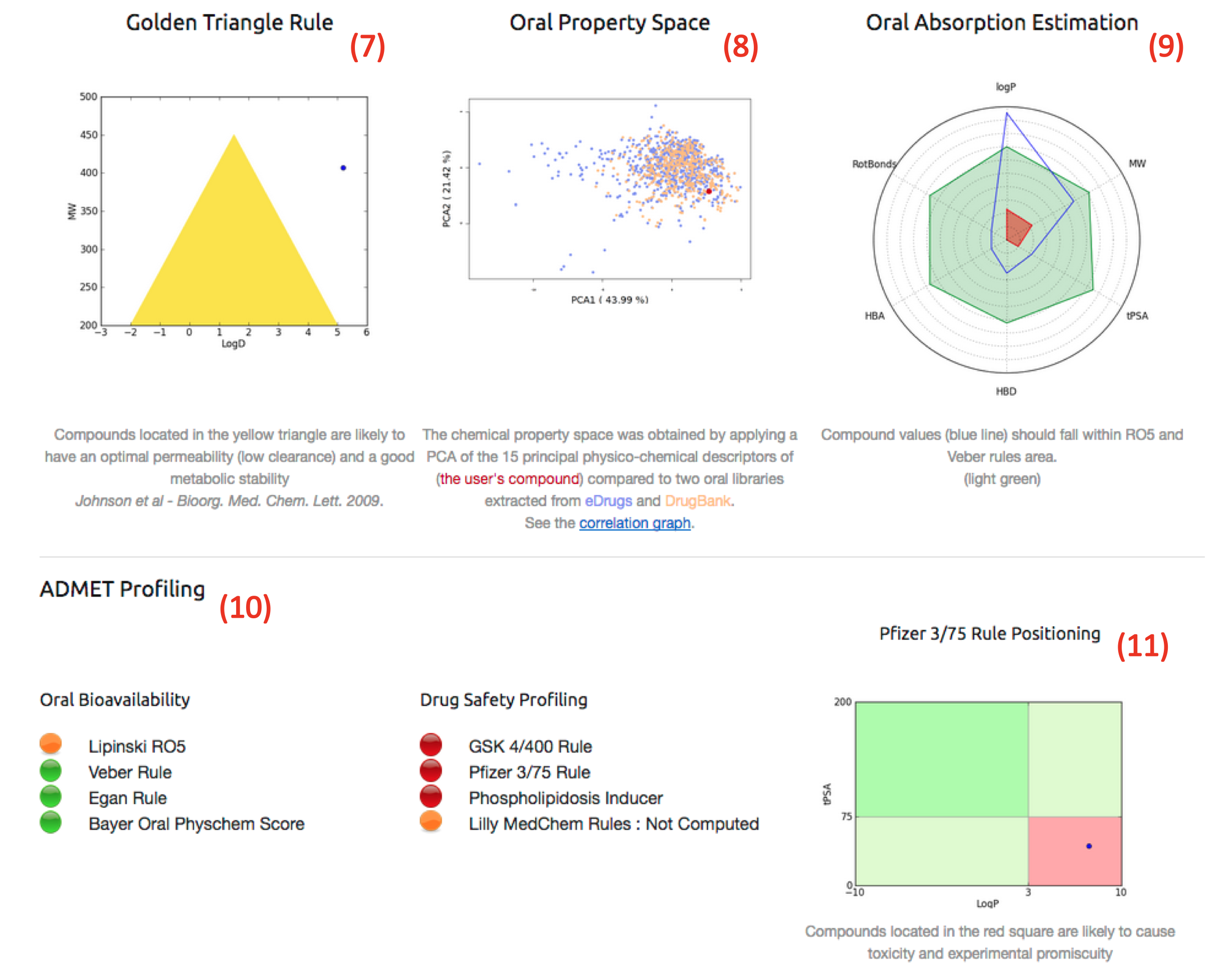
- Regarding the “web-based“ analysing procedure, after having click on the full screen view button (3), the "FAFDrugsHTMLReport" which is divided in 2 sections could by analysed:
Note: All pictures can be enlarged by clicking it.